
- Acne
- Actinic Keratosis
- Aesthetics
- Alopecia
- Atopic Dermatitis
- Buy-and-Bill
- COVID-19
- Case-Based Roundtable
- Chronic Hand Eczema
- Chronic Spontaneous Urticaria
- Drug Watch
- Eczema
- General Dermatology
- Hidradenitis Suppurativa
- Melasma
- NP and PA
- Pediatric Dermatology
- Pigmentary Disorders
- Practice Management
- Precision Medicine and Biologics
- Prurigo Nodularis
- Psoriasis
- Psoriatic Arthritis
- Rare Disease
- Rosacea
- Skin Cancer
- Vitiligo
- Wound Care
Publication
Article
Dermatology Times
New Hope for Treating the 4 Main Types of Epidermolysis Bullosa
Author(s):
Recent advancements in the understanding of epidermolysis bullosa have resulted in a fine-tuning of the classification of the disease group as well as the development of new possible therapies.
Epidermolysis bullosa (EB) is an inherited, heterogenous group of rare genetic disorders characterized by blistering of the skin and mucosal fragility following minimal mechanical trauma and heat.1 Recent advancements in the understanding of EB have resulted in a fine-tuning of the classification of the disease group as well as the development of new potential therapies.
The 4 Main Types of EB
In the United States, the incidence of EB is estimated at 19.57 in 1 million live births, with a prevalence of 11.07 per 1 million population.1 The blistering lesions seen in EB can also occur spontaneously and are the result of the mutations in genes that encode basement membrane zone components of proteins that are responsible for maintaining the integrity of the basal membrane zone and adjacent keratinocytes.



The 4 main types of EB include EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB), and Kindler EB (KEB), the rarest form of the disease. More commonly occurring in the adult population, EB acquisita—a nongenetic type of EB—is an autoimmune disease caused by the immune system erroneously attacking type VII collagen, a key protein in the skin. Significant multiorgan involvement can also occur in the more severe forms of EB, with blisters developing in the mouth, esophagus, stomach, intestines, upper airway, bladder, and genitals; life expectancy here ranges from early infancy to approximately age 30 years, underscoring the urgency for viable treatment options.
EBS is the most common form of EB, occurring in approximately 70% of cases, and is usually caused by mutations in the genes KRT5 and KRT14, which hold the instructions to produce keratin 5 and keratin 14, respectively. JEB is most often caused by mutations in the genes LAMA3, LAMB3, and LAMC3, which hold the instructions to produce laminin 332, or the gene COL17A1, which holds the instructions to make collagen 17. DEB, the autosomal recessive subtype, is considered one of the most severe forms of the disease and is caused by mutations in the gene COL7A1, which holds the instructions to produce collagen VII. KEB, the rarest type of EB with only 250 cases reported worldwide, is caused by mutations in the gene FERMT1, which holds the instructions needed to produce the protein kindlin-1.
The division of these variants is based on the morphological level of separation within the dermal-epidermal junction zone and reflects the underlying molecular abnormality. In EBS, the blisters form within the epidermis; in JEB, they form within the lamina lucida of the basement membrane; in DEB, they form just below the basement membrane; and in KEB, there is a mixed skin cleavage pattern and blisters can occur at all levels of the skin.
“Epidermolysis bullosa simplex represents the mildest form of EB, and many of the patients with EBS will tend to get better later on in life,” said Amy S. Paller, MD. “However, the recessive dystrophic variant of EB is the most urgent, and most eyes are on this form. Patients typically survive the infantile period and continue to get worse in their manifestations, with progressive scarring and tissue damage and, ultimately, aggressive skin cancer. Although all variants are pressing in terms of our search for effective treatment and management options, DEB is so destructive that it remains the most compelling variant.” Paller is chair of the Department of Dermatology and a professor of pediatrics at Northwestern University Feinberg School of Medicine in Chicago, Illinois.

tibanna79/AdobeStock
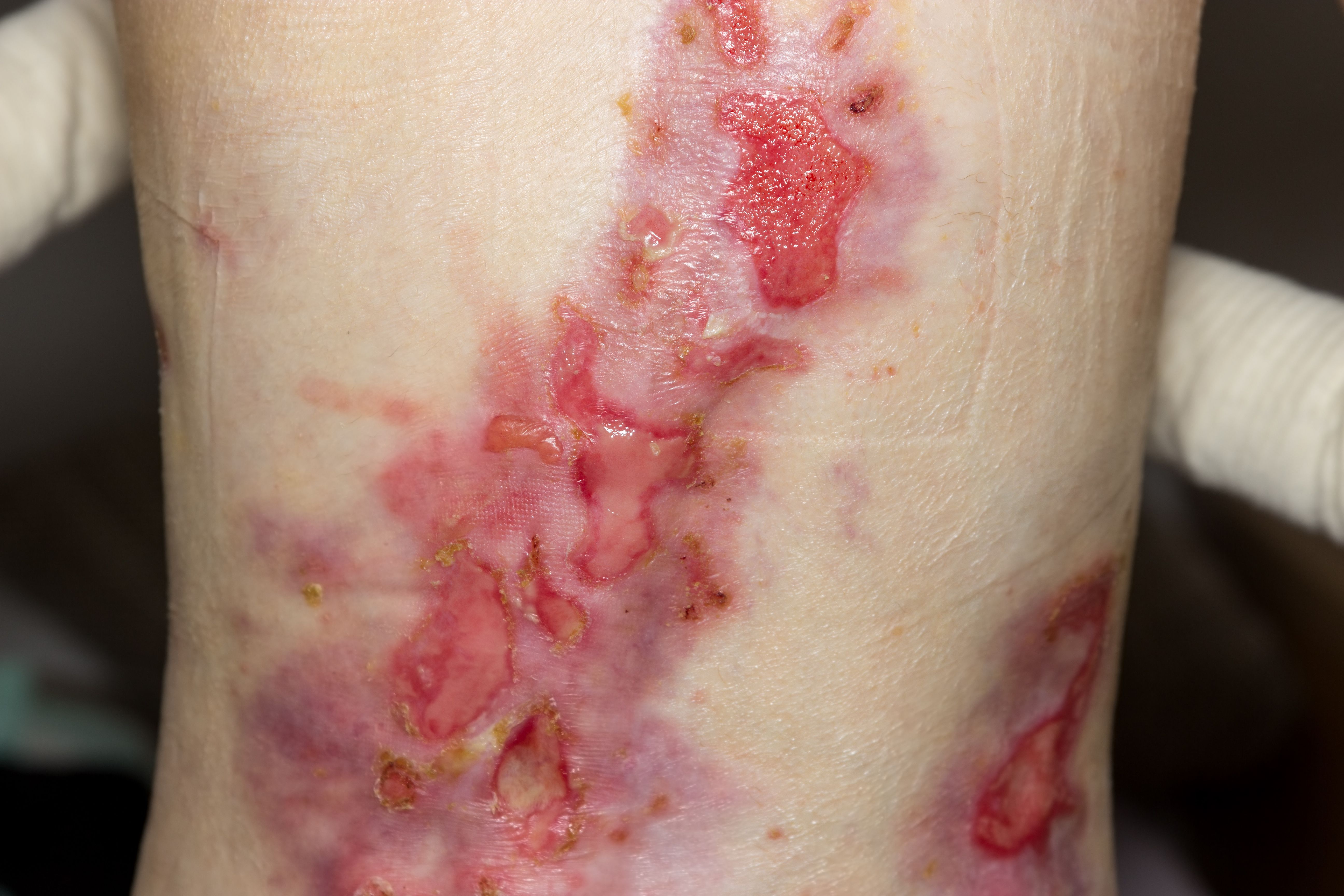
Continued research has resulted in the recognition of more than 30 subtypes of the disease, which are also now categorized under the general umbrella term of EB. According to Paller, the recently updated criteria distinguish between the major EB types and subtypes according to the level of cleavage, inheritance pattern, mutated gene, and the targeted protein. The importance of the revised classification of EB is also in the distinction between classical EB and other skin disorders, in which blister formation is only a minor part of the whole clinical picture, such as peeling skin disorders, erosive disorders, hyperkeratotic disorders, and connective tissue disorders associated with skin fragility.2
Treatment Is Lacking
Unfortunately, few effective therapies are currently available for patients with EB. Treatment and management options are largely supportive and centered on wound care, mitigation of pain and itch, infection control, nutritional support, and prevention and treatment of the associated complications of EB.
“In the absence of curative treatment, multidisciplinary care is aimed at minimizing the risk of blister formation, wound care, physiotherapy management, symptom relief, and detection of other complications—the most feared of which is squamous cell carcinoma...one of the leading causes of mortality in [patients with EB],”Paller said.
Traditionally, the diagnosis of EB could be achieved through biopsy and using immunofluorescence mapping or transmission electron microscopy. Today, however, diagnosis is made through genetic testing, thanks to next-generation gene-mapping sequencing techniques.
According to Paller, researchers and clinicians are employing panels of 28 genes that can be involved in disorders in which blistering is present in the skin of newborns. Using this newer diagnostic technology, clinicians and their patients can expect the results from genetic tests within 4 weeks or sooner. One of the many benefits of genetic testing is that patients with EB can receive more personalized care and earlier medical intervention to minimize trauma and optimally treat blisters and their sequelae. It can also help more quickly identify those patients who may be eligible for clinical trials and newer therapies.
Preclinical or clinical testing of gene therapies (gene replacement, gene editing, RNA-based therapy, natural gene therapy), cell-based therapies (fibroblasts, bone marrow transplantation, mesenchymal stromal cells, induced pluripotential stem cells), recombinant protein therapies, and small molecule and drug repurposing approaches are all being currently explored. According to Paller, these fledgling therapies are generating new hope for an improved patient care and clinical management strategies.
Investigating New Therapies
Ongoing research has led to an increasing number of budding treatment avenues, most of which are still in clinical trials, including topical, oral, and injection modalities.
In May, the FDA approved Krystal Biotech’s beremagene geperpavec (B-VEC; Vyjuvek) for the treatment of DEB.3 The approval was based off of positive data from 2 placebo-controlled clinical trials, GEM3 (NCT04491604) and GEM 1/2 (NCT03536143). B-VEC is a non-invasive topical, redosable gene therapy created to administer 2 copies of the COL7A1 gene when directly applied to DEB wounds.
“The beauty of this drug is its topical application, so it causes no pain and can be implemented even in the youngest children. We are very hopeful that this treatment modality will soon be added to our armamentarium,” Paller said.
Oleogel-S10 (Filsuvez) is also showing promise in patients with JEB and DEB. The birch bark extract rich in betulinis thought to promote wound healing by controlling the inflammatory response and enhancing the mitigation of keratinocytes. Already approved for EB in Europe, the topical gel has met its primary end points in double-blind randomized trials, and according to Paller, has potential to receive FDA approval in the very near future.
Another therapy being investigated in clinical trials is EB-101, consisting of a correct copy of the COL7A1 gene packaged into a retrovirus, which is used to transfer it into the patient’s keratinocytes that were cultured and grown ex vivo. The growing keratinocytes form sheets that serve as skin grafts, expressing the functional type VII collagen protein.
“With just two 8-mm biopsies, you can grow and correct the keratinocytes and create grafts to generate enough product to give the patient enough supply to cover their erosions. This genetically engineered skin graft approach is looking very promising, particularly for larger surface areas of EB-affected skin, such as in those patients with autosomal recessive DEB,” Paller said.
Other gene therapies in development for recessive DEB include gene-corrected fibroblasts from the patient. After introducing the corrected gene to defective cells, the cells are injected every 3 to 6 months into the skin, where they are expected to produce new collagen VII and prevent blister formation. The preliminary trial outcomes appear to yield long-lasting results.
The systemic administration of the broad-spectrum antibiotic gentamicin has also been investigated in clinical trial in infants with recessive DEB and generalized JEB, the results of which showed a positive impact on skin fragility. Some success has been achieved with this investigative therapy, and results look promising, Paller said, although toxicity from the drug still may be of concern.
Drug repurposing in EB has also been explored with dupilumab (Dupixent), an FDA-approved agent for atopic dermatitis. Although it is early, Paller said evidence from ongoing clinical trials show the drug could be helpful in the treatment of the rare EB pruriginosa variant and perhaps other forms.
Hope on the Horizon
This review is a mere snapshot of many novel treatments that are in various stages of development, clinical trials, and approval.
Paller said if all goes well, the next few years may be witness to several effective treatments, which is a very promising outlook for a disease that largely could only be treated symptomatically.
“We are finally being able to start looking at pathogenesis-directed approaches for these diseases as we learn more about not just the underlying genetic causes but also some of the pathways involved in the phenotypes that we see,” she said. “And I think that hopefully over the next 10 years, we are going to have some very good treatments to combine for some of these patients to improve their quality of life.”
Disclosure
Paller is an investigator for Krystal Biotech Inc and Regeneron Pharmaceuticals Inc; a consultant for Aegerion Pharmaceuticals Inc, Castle Creek Biotech Inc, Krystal Biotech Inc, Regeneron Pharmaceuticals Inc, Sanofi Genzyme, and TWi Biotechnology Inc; and on the data safety monitoring boards of Abeona Therapeutics Inc. and InMed Pharmaceuticals.
References
- Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the national epidermolysis bullosa registry. JAMA Dermatol. 2016;152(11):1231-1238. doi:10.1001/jamadermatol.2016.2473
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614-627. doi:10.1111/bjd.18921
- FDA approves first topical gene therapy for treatment of wounds in patients with dystrophic epidermolysis bullosa. U.S. Food and Drug Administration. May 19, 2023. Accessed May 19, 2023. https://www.fda.gov/news-events/press-announcements/fda-approves-first-topical-gene-therapy-treatment-wounds-patients-dystrophic-epidermolysis-bullosa.
















Newsletter
Like what you’re reading? Subscribe to Dermatology Times for weekly updates on therapies, innovations, and real-world practice tips.













